5 min read
In the pharmaceutical industry, every part of the production process – from the amount of each ingredient included in a batch to the cleaning process between batches – must be documented. This documentation becomes an important part of a product’s genealogy, which means it must be stored long term and easy to produce when required by a regulatory authority. But this documentation is really only as good as the data collected to produce the documentation. This means data integrity, which focuses on providing and maintaining the accuracy and consistency of data over its entire lifecycle, is central to compliance in this heavily regulated industry.
Understanding Data Integrity for Pharmaceutical Production
At a high-level, data integrity is all about the completeness, consistency, and accuracy of data. As data is generated and collected, you need to be sure it is good data, and you must know who was responsible for the actions that led to the data generation and collection. Data also needs to remain complete, consistent, and accurate throughout the entire process, which includes creation, modification, processing, maintaining, archiving, retrieving, and final disposition of the data. Another key aspect to ensuring data integrity is that you must have good documentation practices (GDPs) in place for recording and storing the data.
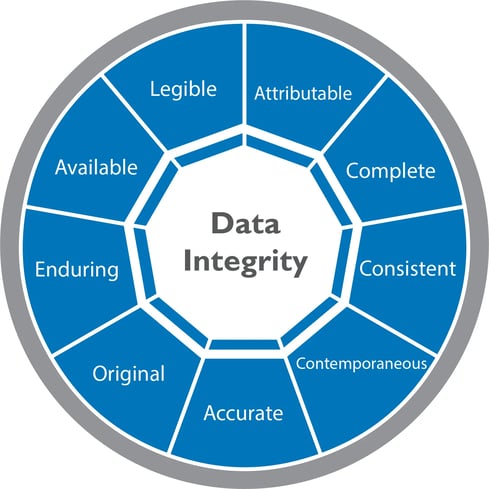
For the pharmaceutical industry, the United States Food and Drug Administration (FDA) first established a set of principles that serve as the cornerstone for ensuring GDPs and data integrity in the 1990’s. Known as ALCOA, and later becoming ALOCA+ in the 2010s, these principles include the following attributes:
- Attributable – Clearly show who observed and recorded data
- Legible – Easy to understand and permanent
- Contemporaneous – Recorded as it was observed when it was executed and not retrospectively
- Original – Accessible and preserved in native form
- Accurate – Error-free and conforming with protocols
- Complete – Include an audit trail that shows nothing was lost or deleted
- Consistent – Displayed the same way wherever it is accessed
- Enduring – Accessible and readable during the entire lifecycle
- Available – Documents should be accessible in a readable format to required personnel
 At Applied Control Engineering (ACE), we have decades of experience working with pharmaceutical companies to ensure data integrity by automating data collection methods and integrating intelligent historians for data storage. Below are our top five recommendations for implementing risk-based strategies to prevent data integrity issues using the key principles outlined within ALOCA+, which will ultimately help your organization meet regulatory requirements.
At Applied Control Engineering (ACE), we have decades of experience working with pharmaceutical companies to ensure data integrity by automating data collection methods and integrating intelligent historians for data storage. Below are our top five recommendations for implementing risk-based strategies to prevent data integrity issues using the key principles outlined within ALOCA+, which will ultimately help your organization meet regulatory requirements.
Recommendation 1: Electronic Record Keeping in Compliance with 21 CFR Part 11
Historically, the pharmaceutical industry has included a variety of manual processes and handwritten data recording methods, which is a huge risk to data integrity as this presents a lot of opportunity for human error. One key driver to move more pharmaceutical companies to electronic record keeping occurred in the 1990s when the FDA adopted regulation 21 CFR Part 11, which defines requirements for electronic records and electronic signatures (ER/ES).
In short, 21 CFR Part 11 requires all electronic signers to prove their identity every time, which may be done by entering a password or using some biometric identifier. Part 11 also requires an audit trail of every signature and the action/event/change that was signed for. To further reduce risk by providing an even higher level of trust in ER/ES, we work with customers to design electronic signature systems using key security principles for IT business systems including enforcing rules for password complexity, requiring regular password changes, and implementing two-factor authentication.
Recommendation 2: Proper Access Control
Access control is not as simple as having a single password for all users to login to an operator station. Instead, access control should include a combination of computerized and procedural controls. Every user should have a unique login with access permissions based on clearly defined roles within the organization. Not every user needs the same level of access to all systems. As an example, an operator that has access to one area of the process should be limited or have no access to areas of the process in which they are not trained.
To be sure data is attributable, the access control system should authenticate all users and maintain a record of who logged into the system, where the login occurred, and when the login occurred as well as data on who granted approvals. Additionally, to ensure data is contemporaneous and original, controls should be put into place to make sure data is not lost or obscured in any way and that every activity someone takes is documented at the time the activity occurs, not later.
Recommendation 3: Create an Audit Trail
To reduce the risk of having incomplete data, current good manufacturing practices (GMPs) require audit trails that maintain a secure, time-stamped record of all data creation, modification, or deletion. An alert triggering an audit trail should occur every time a change is made to the system. For example, if a validated production recipe calls for an agitator to run at 100 RPMs, but the setpoint is changed to 105 RPMs during the execution of that recipe, the system should require the operator to complete an electronic form to record the old and new value, the date and time the change was made, who authorized the change, and possibly a comment as to the reason why the change was made. These electronic audit forms should also be available in a batch report so that all changes can be easily reviewed, if necessary.
Recommendation 4: Verification of Manual Processes
While many systems and processes involved in pharmaceutical production have been automated, there are still some manual processes involved. For example, if manually added ingredients going into a batch must be weighed, the operator may have to manually enter the weight before the batch starts. When a validated electronic means of capturing weight is not available to ensure the weight is entered properly, it should be verified by a second person such as the shift supervisor. This secondary verification should require the user to enter their unique username and password to verify the weight, which will help minimize data integrity risks and ensure the reliability and accuracy of manual data.
Recommendation 5: Easily Accessible, Readable Data
Since regular reporting is required to demonstrate compliance, you need a system for organizing and easily viewing data in a human-readable format. ACE can work with your team to aggregate the data from your systems into reports and dashboards so that you can run your plant and meet regulatory requirements. We can also help identify retention policies, meet traceability requirements, and develop processes for storing data for long-term access and ease of retrieval.
Learn more about our experience working in the pharmaceutical industry and our expertise executing data collection and storage systems that will reduce data integrity risks.
Related Posts
The Future of OT Data Integration: Trends, Challenges, and Opportunities
Mankind has always advanced by improving how we collect, process, and share information. Today,...
Driving Efficiency and Visibility with Real-Time Production Tracking
In today's fast-paced manufacturing landscape, staying ahead of the competition requires efficient...
How to Communicate Modbus Natively with ControlLogix
The Modbus protocol was originally published in 1979 by Modicon as a simple request-response...